Docking studies on protein targets for diabetes mellitus II
Assistant researcher project in computational and medicinal chemistry (2016 - 2017)
University of Chile, Faculty of Chemical and Pharmaceutical Science
Diabetes mellitus type 2 (DMT2) is a metabolic disease where chronically increase the glycemia on patients with pancreatic insulin secretion failure, insulin resistance, and stimulation of the synthetic glucose pathway in the liver. To treat DMT2, several enzymes involved in glycemic control, such as glycogen phosphorylase A (GPa), α-(1,4)-glucosidase and tyrosine phosphatase-1B (PTP-1B) have been studied as protein targets. Even though acarbose (Glucobay®) is the most common anti-hyperglycemic drug used for the treatment of DMT2 displaying a good efficiency in attenuating the rise of glucose levels in plasma it has been associated with undesirable side effects. As an alternative it was found that coumarins display α-(1,4)-glucosidase inhibition. Coumarins are molecules formed by a benzene ring fused to an α-pyrone. These compounds have been highly studied due to their diverse reported pharmacological activities, depending on the functional groups attached to the scaffold.
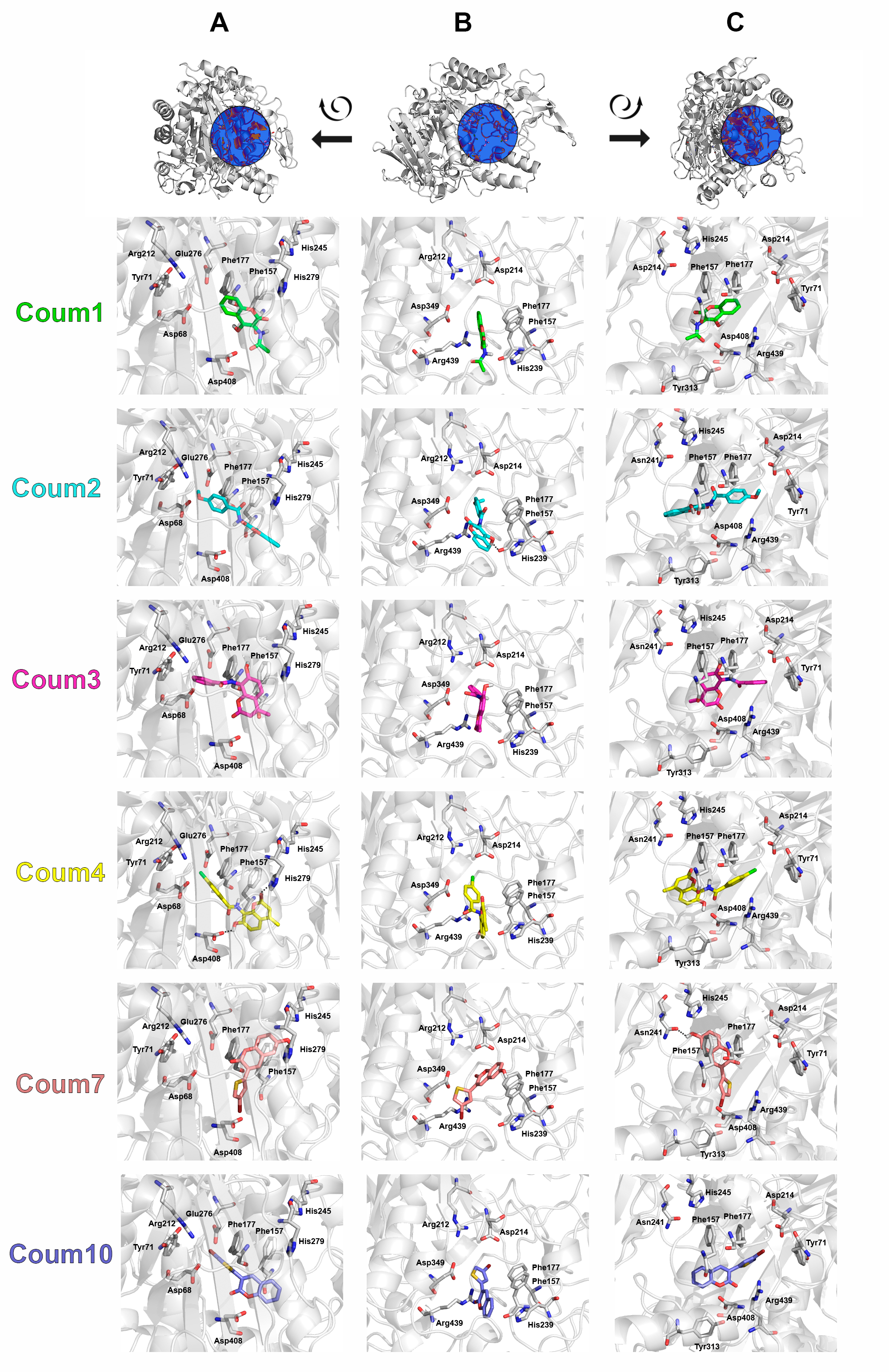
In this project in collaboration with experimental scientists from the University of Chile, I investigated the union of different coumarins derivatives on GPa, PTP-1B and alpha-(1,4)-glucosidase. The aim of this work was to determine both the inhibitory potency and type of inhibition of a series of new synthesized coumarins (bearing methoxy, hydroxy, amide and aryl groups in different positions).
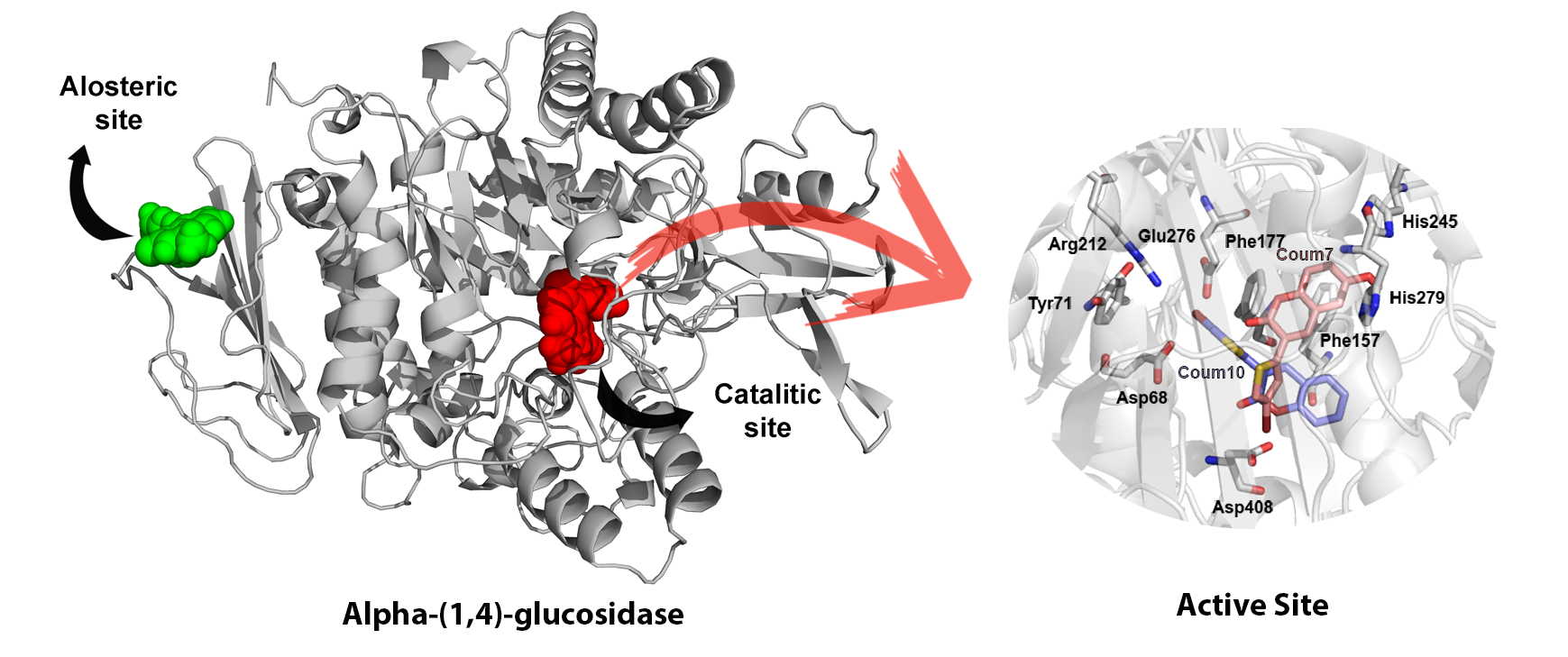
The computational study was carried out to identify and compare the catalytic site or a second hydrophobic site to better understand the interactions between coumarins and the enzyme under study. Due to the lack of structural information of α-(1,4)-glucosidase homology models were obtained using the Swiss model server and sequences from UniProt Server. Then BLAST analysis was performed to obtain protein templates to create the protein models. The coumarin studied were created and optimised using Gaussian09 and charge parameters were obtained using RED-III.4 program. Finally, docking calculations were obtained using Autodock Vina and the results were analysed using energy scoring values and interactions using LigPlot+. Our results published in *Current Topics in Medicinal Chemistry* revealed that coumarins bind in a more external area of α-(1,4)-glucosidase comparing to the substrate displaying aromatic and hydrophobic interactions, as well as some hydrogen bonds. Aromatic interactions with two phenylalanine residues, 157 and 177, were the most common among the studied coumarins.