Simulation Workflow projects
Computational Scientist, Chemistry (2017-2022)
Institute of Nanotechnology, Karlsruhe Institute of Technology (Germany)
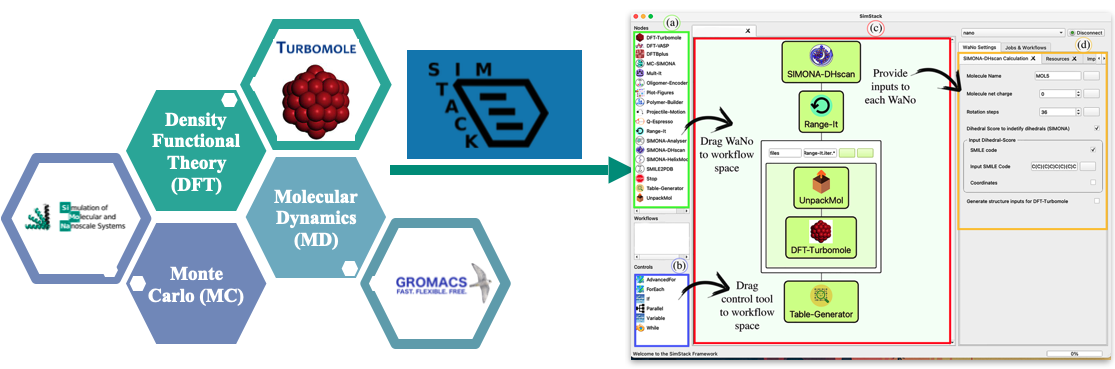
SimStack is a lightweight client-server program developed by Prof. Wenzel’s group at the Karlsruhe Institute of Technology (KIT) and Nanomatch GmbH. It provides a graphical user interface to construct simulation workflows from individual program or protocol units called Workflow Active Nodes (WaNos) using High Power Computer (HPC) resources. SimStack uses Extensible Markup Language (XML) format to generate the graphical interface for the settings of each WaNo, which is human readable and user friendly language, even for non-developer users. SimStack can be easily used by non-computer scientists to make closer the relationship between experiment-theory and vice versa.
Project 1
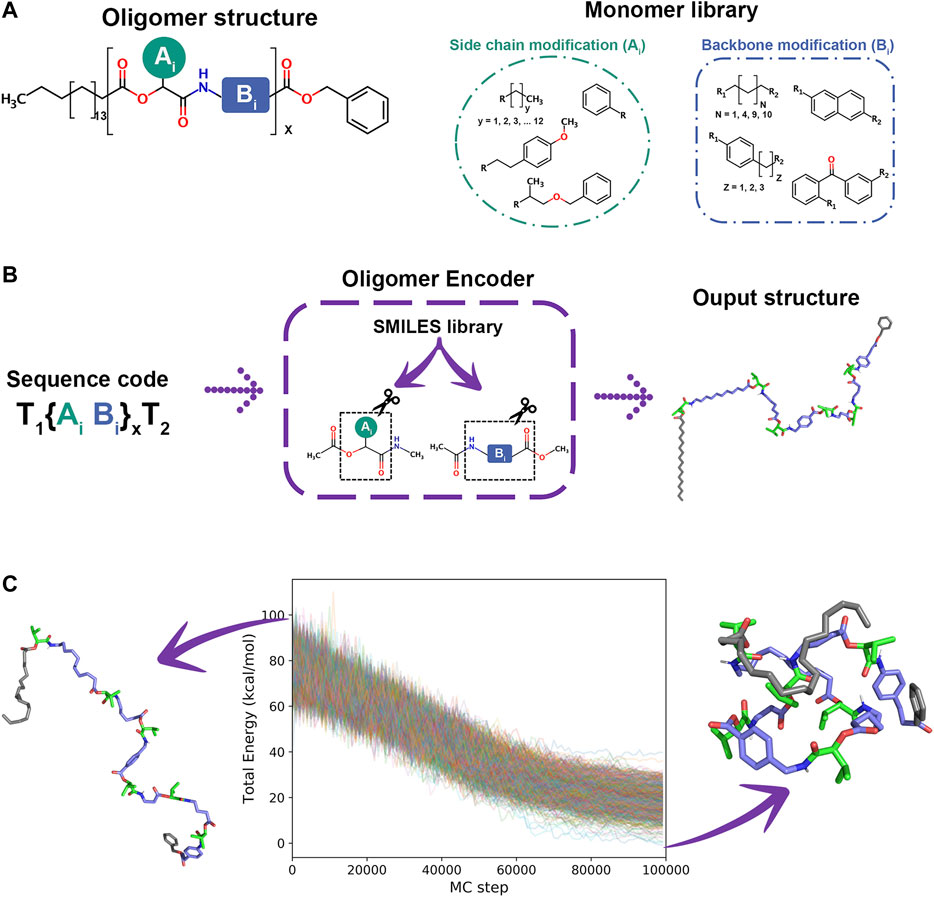
SimStack was used to study the possible conformations of oligomer sequences. A protocol was created to systematically create oligomer atomic models from Oligomer Encoder (OE) sequences. OE is a tool containing a monomer library of SMILES codes linked to a three letter code similar to a 3-letter proteins amino acid system. These monomers are then optimised using TURBOMOLE . The optimized monomer structures are used to calculate charges using the AM1-BCC method. Parameters from the General Amber FF are extracted with Acpype to generate GROMACS input files. Once GROMACS input files are created, Monte Carlo simulation inputs are generated using the Python protocol provided by SIMONA. This strategy allows OE to be a highly scalable molecule builder.
Publications
Project 2
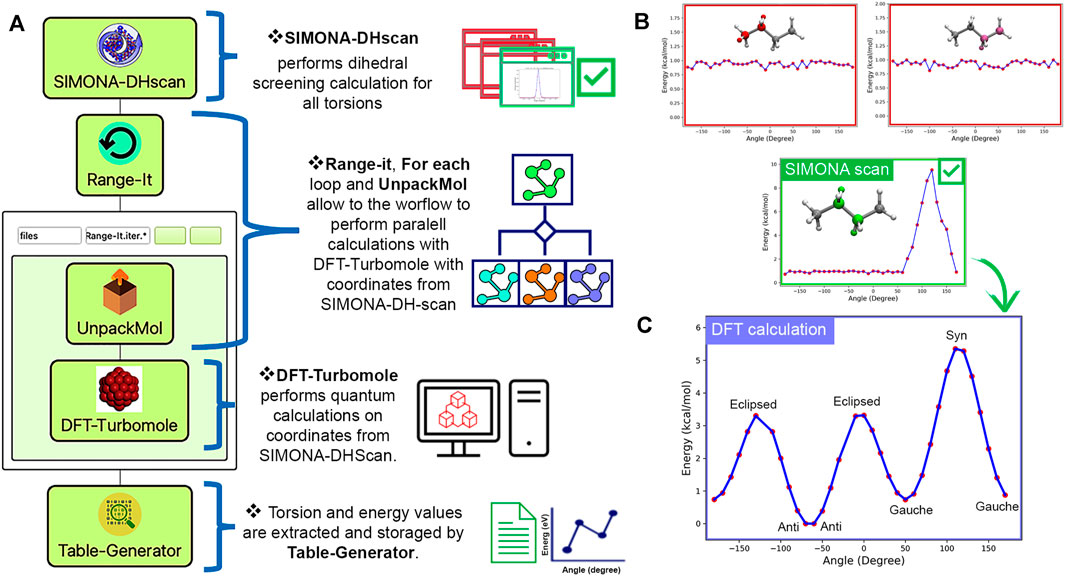
SimStack was also used to explore the possible dihedral values on the backbone of the polymer, using SIMONA, TURBOMOLE, and SimStack. The WaNo SIMONADHscan was built to generate molecular structures using SMILES code as input. Then a specific SIMONA scan protocol was individually applied to all possible torsion angles identified. The scan protocol uses the metropolis MC algorithm to perform arbitrary rotations of a selected torsion angle and apply relaxations on adjacent torsion angles. The total energy of each configuration is calculated using the Coulomb, and Lenard-Jones terms from GAFF.